Deep learning architectures such as CNNs and transformers have significantly advanced biological sequence modeling by capturing local and long -term dependencies. However, their use in biological contexts is limited by high calculation requirements and the need for large data sets. CNNs effectively detect local sequence patterns with subquadratic scaling, whereas transformers utilize self -storage to model global interactions, but require square scaling, making them expensive expensive. Hybrid models, such as Enformers, integrate CNNs and transformers to balance local and international context modeling, but they still face scalability problems. Large -scale transformer -based models, including Alphafold2 and ESM3, have achieved breakthroughs in predicting protein structure and sequence function modeling. Nevertheless, their dependence on extensive parameter scaling limits their efficiency in biological systems, where data availability is often limited. This highlights the need for more calculation-efficient approaches to model-to-function ratio exactly.
To overcome these challenges, the epistasis – the interaction between mutations within a sequence – provides a structured mathematical framework for biological sequence modeling. Multilinear polynomes can represent these interactions and offer a principled way of understanding sequence function relations. State Space Models (SSMS) is naturally in line with this polynomial structure using hidden dimensions to approach epistatic effects. Unlike transformers, SSMS uses fast Fourier Transform (FFT) Conventions to model global dependencies effectively while maintaining subquadratic scaling. In addition, the integration of gated depth -cohabitation of local extraction of function and expressiveness improves through the choice of adaptive function. This hybrid approach balances calculation efficiency with interpretability, making it a promising alternative to transformer-based architectures for biological sequence modeling.
Researchers from institutions, including MIT, Harvard and Carnegie Mellon, introduce Lyra, a subquadratic sequence modeling architecture designed for biological applications. LYRA integrates SSMs to capture long -range dependencies with projected fenced -in interventions to local function extraction, enabling effective o (n log n) scaling. It effectively models epistatic interactions and achieves advanced performance across over 100 biological tasks, including protein fitness prediction, RNA function analysis and CRISPR GUIDE DESIGN. Lyra operates with significantly fewer parameters – up to 120,000 times less than existing models – while they are 64.18 times faster in the inference and democratizing access to advanced biological sequence modeling.
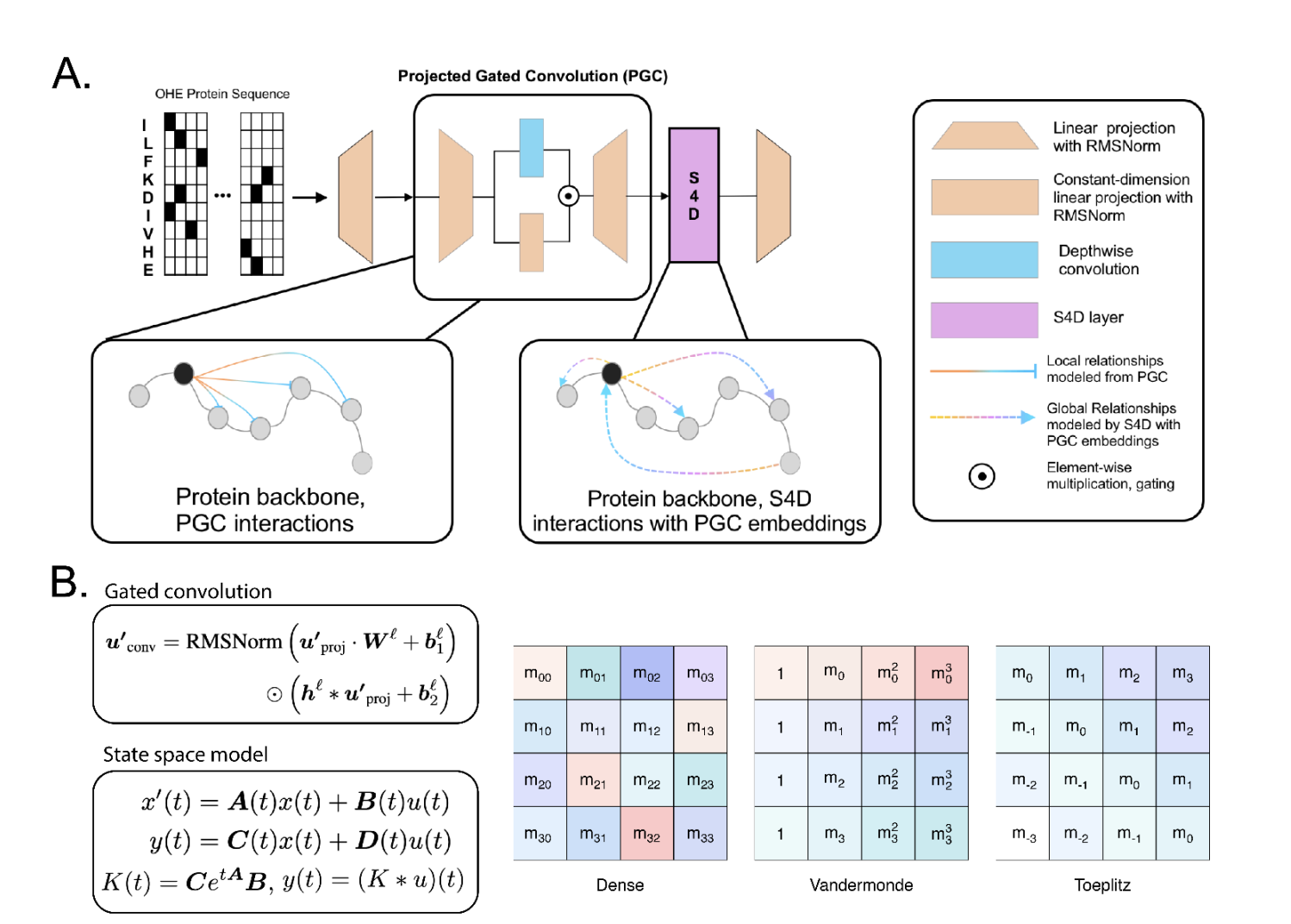
Lyra consists of two key components: Projected Gated Convolution (PGC) blocks and a state -stroke with depth convolution (S4D). With approximately 55,000 parameters, the model includes two PGC blocks for recording local dependencies, followed by an S4D layer for modeling long-range interactions. PGC processes input sequences by projecting them for intermediate dimensions, applying depth 1D cohabitation and linear projections and recombining functions through element-wise multiplication. S4D utilizes diagonal state space models to calculate convolution kernels using matrixes A, B and C, which effectively captures sequence breaks through weighted exponential terms and improves Lyra’s ability to model biological data effectively.
Lyra is a sequence modeling architecture designed to capture local and long -range dependencies in biological sequences effectively. It integrates PGCs for localized modeling and diagonalized S4D into global interactions. Lyra approaches complex epistatic interactions using polynomial expressiveness that surpass transformer -based models in tasks such as protein fitness country scape prediction and deep mutation scanning. It achieves advanced accuracy across different protein and nucleic acid modeling uses, including disturbance prediction, mutation effect analysis and RNA dependent RNA polymerasis detection, while maintaining a significantly smaller parameter number and lower calculation costs than existing large models.
Finally, Lyra introduces a subquadratic architecture for biological sequence modeling that utilizes SSMs to approximate multilinear polynomial functions effectively. This enables superior modeling of epistatic interactions while significantly reducing calculation requirements. By integrating PGCs into local functional extraction, LYRA achieves advanced performance across over 100 biological tasks, including protein fitness prediction, RNA analysis and CRISPR guide design. It surpasses large foundation models with far fewer parameters and faster inference, which requires only one or two GPUs for training within a few hours. Lyra’s effectiveness democratizes access to advanced biological modeling with therapeutics, pathogen surveillance and bioproduction applications.
Check out the paper. All credit for this research goes to the researchers in this project. You are also welcome to follow us on Twitter And don’t forget to join our 85k+ ml subbreddit.
Sana Hassan, a consultant intern at MarkTechpost and dual-degree students at IIT Madras, is passionate about using technology and AI to tackle challenges in the real world. With a great interest in solving practical problems, he brings a new perspective to the intersection of AI and real solutions.